Selective articles about NERDSS
We kindly ask that you cite: Varga, M., Fu, Y., Loggia, S., Yogurtcu, O.N., & M.E. Johnson NERDSS: a nonequilibrium simulator for multibody self-assembly at the cellular scale. Biophysical Journal 118, P3026-P3040 (2020) in all publications that make use of NERDSS.
Below are selective articles that made use of NERDSS:
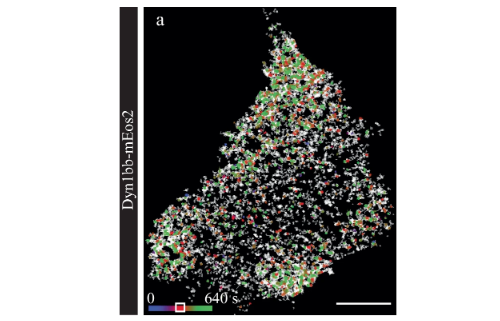
Dynamin1 long-and short-tail isoforms exploit distinct recruitment and spatial patterns to form endocytic nanoclusters
Jiang, Anmin, et al. Nature Communications 15.1 (2024)
Endocytosis requires a coordinated framework of molecular interactions that ultimately lead to the fission of nascent endocytic structures. How cytosolic proteins such as dynamin concentrate at discrete sites that are sparsely distributed across the plasma membrane remains poorly understood. NERDSS is used to model the 3D to 2D binding of dynamin to the cell membrane and assess the binding kinetics of the reaction.
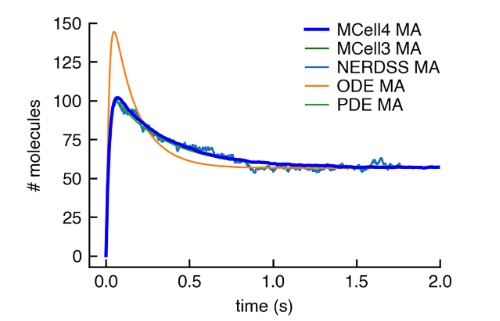
Mcell4 with bionetgen: A Monte Carlo simulator of rule-based reaction-diffusion systems with python interface
Husar, Adam, et al. PLOS Computational Biology 20.4 (2024)
The authors describe and validate a new version of the open-source MCell simulation program (MCell4), which supports generalized 3D Monte Carlo modeling of diffusion and chemical reaction of discrete molecules and macromolecular complexes in solution, on surfaces representing membranes, and combinations thereof. For volume-surface and surface-surface reactions, MCell subdivides the surface areas of geometric objects into small tiles with a maximum of one molecule can occupy one tile at a time. Simulations that involve binding to a surface are performed with MCell4 and with NERDSS, MCell3, and VCell. Their results show good alignment and the algorithm that MCell4 incorporates is validated.
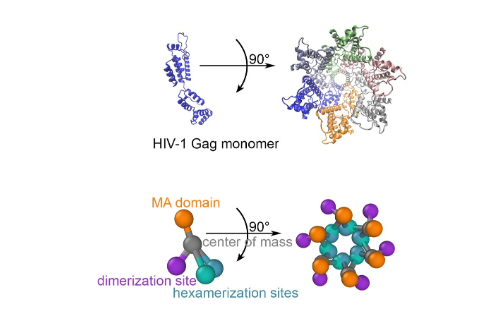
Structure of the HIV immature lattice allows for essential lattice remodeling within budded virions
Guo, Sikao, et al. elife 12 (2023)
For HIV virions to become infectious, the immature lattice of Gag polyproteins attached to the virion membrane must be cleaved. Cleavage cannot initiate without the protease formed by the homo-dimerization of domains linked to Gag. However, The mechanism of Gag-Pol dimerization is unknown. The authors use NERDSS to carry out simulations of the immature Gag lattice as derived from experimental structures, showing that dynamics of the lattice on the membrane is unavoidable due to the missing 1/3 of the spherical protein coat. These dynamics allow for Gag-Pol molecules carrying the protease domains to detach and reattach at new places within the lattice.
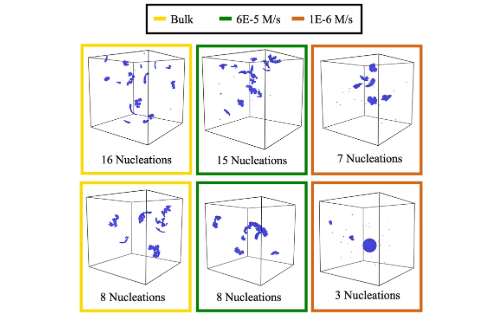
Temporal control by cofactors prevents kinetic trapping in retroviral Gag lattice assembly
Qian, Yian, et al. Biophysical Journal 122.15 (2023)
For retroviruses like HIV to proliferate, they must form virions shaped by the self-assembly of Gag polyproteins into a rigid lattice. The energetic criterion for forming stable lattices is unknown, as are their corresponding rates. Here, the authors use NERDSS simulations with models designed from the cryo-ET structure of the immature Gag lattice to map a phase diagram of assembly outcomes controlled by experimentally constrained rates and free energies over experimentally relevant timescales. It is shown that multiple Gag lattices nucleate before growth can complete, resulting in loss of free monomers and frequent kinetic trapping. Based on the simulations, the authors propose a mechanism by which Gag binding to IP6 can provide the additional time delay necessary to support smooth growth of the immature lattice with relatively fast assembly kinetics and avoid most kinetic traps.
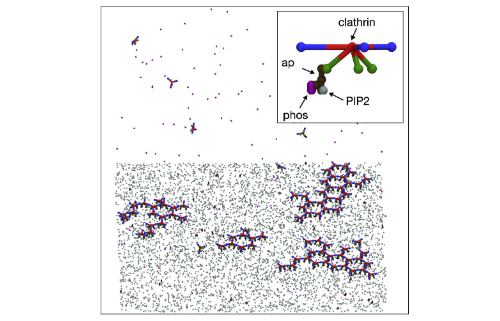
Large self-assembled clathrin lattices spontaneously disassemble without sufficient adaptor proteins
Guo, Sikao, et al. Biophysical Journal 121.3 (2022)
Clathrin-coated structures must assemble on cell membranes to internalize receptors. These structures can grow surprisingly large, containing over 20 clathrin seemingly locked in a hexagonal lattice, yet often fail to form productive vesicles. The authors combine in vitro kinetic measurements and NERDSS simulations to build a theory to differentiate mechanisms of stable vs unstable clathrin assembly on membranes. Their model predicts how adaptor protein density controls stabilization of clathrin-coated structures against spontaneous disassembly, and shows ATPases are not required for lattice remodeling, which is a critical advance towards predicting productive vesicle formation.
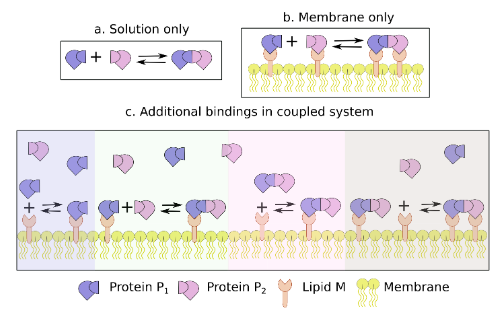
Speed limits of protein assembly with reversible membrane localization
Mishra, Bhavya, et al. J. Chem. Phys. 154, 194101 (2021)
Protein assembly is often studied in a three-dimensional solution, but a significant fraction of binding events involve proteins that can reversibly bind and diffuse along a two-dimensional surface. The authors find that proteins can accelerate dimer formation due to an increase in relative concentration after change in dimensionality, driving more frequent collisions, which often win out over slowdowns due to diffusion. With validations from NERDSS simulations, they derive a single expression for the characteristic timescale of the 3D to 2D assembly process, where the change in dimensionality renders rates and concentrations effectively time-dependent.
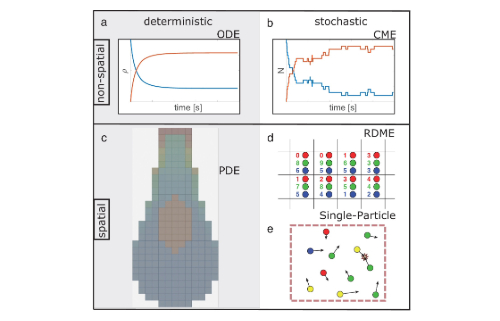
Quantifying the roles of space and stochasticity in computer simulations for cell biology and cellular biochemistry
Johnson, Margaret. E., et al. Molecular Biology of the CellVol. 32, No. 2 (2021)
The authors defined a series of computational test cases ranging from very simple to moderately complex, varying key features of dimensionality, reaction type, reaction speed, crowding, and cell size. A number of commonly used computational programs or approaches including NERDSS were tested on these test cases. The strengths and limitations of these programs and approaches for exploring cell biological questions were discussed to provide a framework for decision making by researchers developing new models.
The algorithm that NERDSS is based of is explained in the following articles:
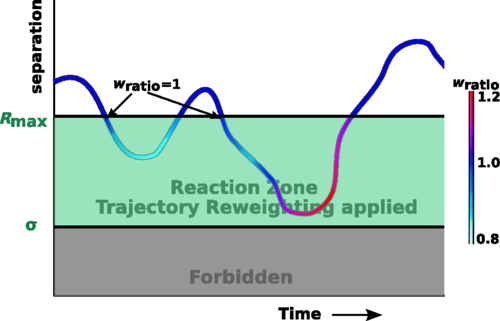
Free propagator reweighting integrator
for single-particle dynamics in reaction-diffusion models of heterogeneous protein-protein interaction systems
Johnson, Margaret E. & Hummer, Gerhard, Phys. Rev. X 4, 031037 (2014).
This paper introduces the free-propagator reweighting algorithm (FPR) for particle-based RD. We combine the use of the exact Green's function for a pair of reacting particles with the approximate free-diffusion propagator for position updates to particles. Trajectory reweighting in the FPR method is able to recover the exact association rates for a pair of interacting particles at all times.
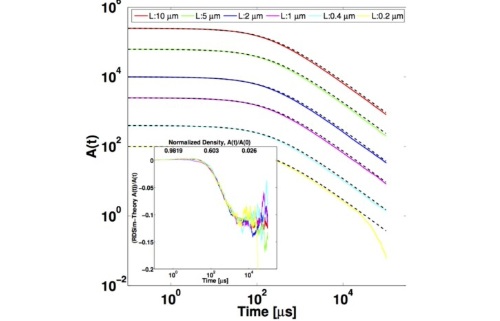
Theory of bi-molecular association dynamics in 2D for accurate model and experimental parameterization of binding rates
Yogurtcu, Osman N. & Johnson, Margaret E., J. Chem. Phys. 143, 084117 (2015)
The dynamics of association between diffusing and reacting molecular species are routinely quantified using simple rate-equation kinetics that assume both well-mixed concentrations of species and a single rate constant for parameterizing the binding rate. In two-dimensions (2D), however, even when systems are well-mixed, the assumption of a single characteristic rate constant for describing association is not generally accurate, due to the properties of diffusional searching in dimensions d ≤ 2. When FPR is extended to 2D, diffusion impacts more strongly the reaction kinetics in these 2D systems.
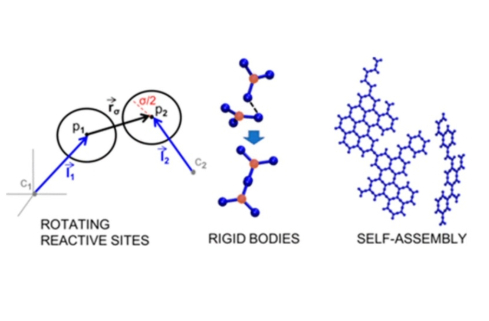
Modeling the self-assembly of protein complexes through a rigid-body rotational reaction-diffusion algorithm
Johnson, Margaret E., J Phys Chem B. 122, 11771 (2018)
We develop a relatively simple but accurate approach for building rigid structure and rotation into single-particle reaction-diffusion methods, providing a rate-based method for studying protein self-assembly. This approach reproduces both the kinetics of association, which is altered by rotational diffusion, and the equilibrium of reversible association, which is not. The macroscopic kinetics of association can also be predicted on the basis of the microscopic parameters of our structurally resolved model, allowing for critical comparisons with theory and other rate-based simulations.
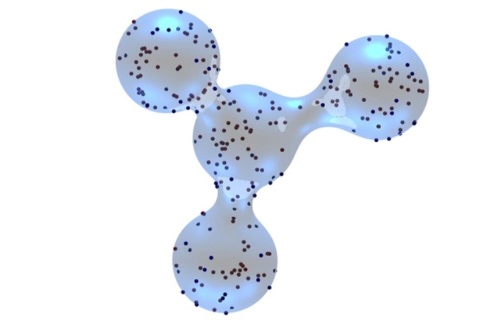
An implicit lipid model for efficient reaction-diffusion simulations of protein binding to surfaces of arbitrary topology
Fu, Yiben, et al., J Chem Phys. 151, 124115 (2019)
This paper introduces an algorithm for reversible binding of proteins to continuum surfaces with implicit lipids, providing dramatic speed-ups to many body simulations. This algorithm implements FPR 3D-to-2D to account for the reactive flux at a surface and handle diffusion on surfaces that have arbitrary topologies.